Chapter 4 Workflow - Step 2
Step 2 completes multiple steps and reports output to one MultiQC report at the end, with various important outputs to the History. The most useful output from this workflow for Differential Expression (DE) analysis is the table of counts with rows as gene names and columns as sample names.
The details of the pipeline are visible by following the workflow URL. In short, it uses trimmomatic, RNA STAR Aligner and featureCounts.
4.1 Import workflow
Follow instructions in Section 3.1 to add the following workflow:
4.2 Run Workflow
There are 4 required inputs to this workflow. Set it up as follows:
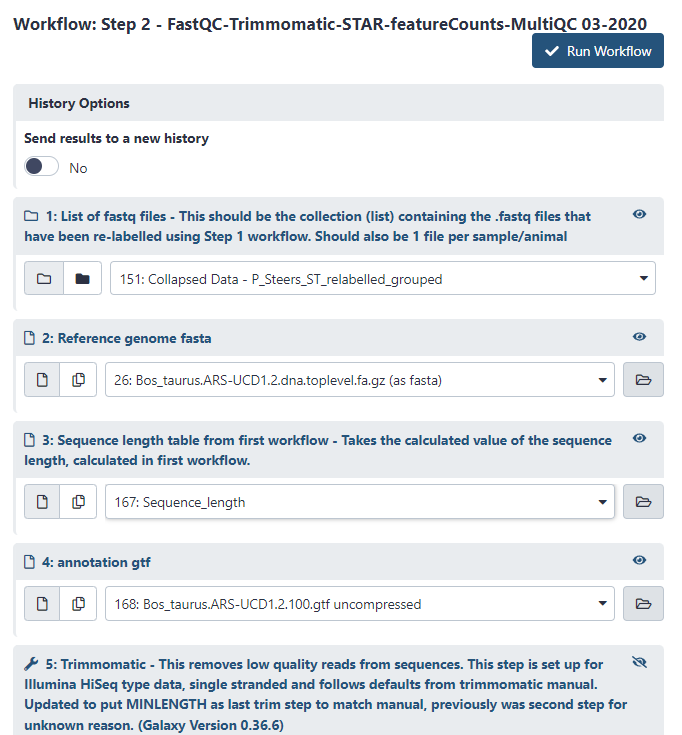
Figure 4.1: Setup Step 2 then press Run Workflow
This workflow will take a very long time to run. It runs on the Galaxy server and you can log in from any computer to see it’s status. You’re computer does not need to stay on. An email will also be sent to you once the last step is completed.
4.3 Save data
Each analysis will require different outputs, but a simple DE analysis will normally only require the featureCounts output that is normally labelled as featureCounts_matrix.tabular. Download this file. It can be previewed in Microsoft Excel by dragging and dropping into an active Excel window, however it is sometimes a large file and best handled in R or other similar software. Also be careful not to save changes if viewing in Excel as some gene names will be converted to date formats in excel and may introduce errors e.g. MARCH1 gene.
It is also recommended to save the MultiQC webpage and stats output. This is particularly useful for describing methods when writing up.
You may also want to view the specific alignments and therefore you will need the .bam files. See https://software.broadinstitute.org/software/igv/BAM for more information.
4.4 Inspect Report
It is important to view the MultiQC webpage and check that all output meets your quality requirements.